
Today’s language of science is Enlgish. As a Swede, that means that certain domains of the Swedish language have either regressed into oblivion, or never developed in the first place. There are so many specific terms for each sub-discipine of science, and I have realized that even some of the core concepts of evolutionary biology lack terms in Swedish, such that Swedish evolutionary biologists talking to each other in Swedish use the English terms.
These are not only my own reflections; I share them and I share the interest for language with my husband, Spoke Wintersparv. As a project to highlight this, he therefore took on the crazy challenge to translate one of my papers in full into Swedish. It was quite the undertaking for us both, but we are happy to present the results below.
Trevlig läsning!
Utredning av den västpalearktiska blåmesens (Cyanistes spp.) komplexa evolutionshistoria – fylogenomiska analyser påvisar radiering genom åtskilliga kolonisationstillfällen med efterföljande isolering
MARTIN STERVANDER,* JUAN CARLOS ILLERA,† LAURA KVIST,‡ PEDRO BARBOSA,* NAOMI P. KEEHNEN,*PETER PRUISSCHER,*STAFFAN BENSCH*and BENGT HANSSON*
*Molekylär ekologi och evolution, Biologiska institutionen, Lunds universitet, Ekologihuset, 223 62 Lund, Sverige, †Forskningsenheten för biologisk mångfald (UO-CSIC-PA), Oviedo universitet, Campus of Mieres, Research Building, 5 vån, Calle Gonzalo Guitérrez Quirós, s/n, 33600 Mieres, Asturias, Spanien, ‡Biologiska institutionen, Oulu universitet, POB 3000, 90014 Oulu, Finland
Sammanfattning
Isolerade öar och deras ofta unika biota fortsätter att spela en central roll i vår förståelse av vikten av genetisk drift, variation och adaption i populationsdifferentierings- och artbildningsprocessen. Ett ösystem som har inspirerat och väckt intresse hos evolutionsbiologer är blåmeskomplexet (Cyanistesspp.) i Europa och Afrika, i synnerhet den komplexa evolutionshistorien för Kanarieöarnas mångsidigt genetiskt utpräglade taxa. Kunskap om afrokanariska kolonisationstillfällen är av särskild vikt, på grund av nya okonventionella förslag om att dessa öpopulationer utgjort källan till den utbredda populationen på den afrikanska kontinenten. Genom att använda en kombination av Sangersekvensering på populationsnivå (20 loci; 12 500 nukleotider) och parallellsekvensering av enstaka representanter för populationer (>3 200 000 nukleotider), analyserade i koalescensbaserade och fylogenetiska ramverk, undersökte vi förhållandet mellan blåmesar från fastlandet, med deras artfränder på Kanarieöarna. Vi fann (i) att afrokanariska blåmesar är monofyletiska och representerar fyra överordnade klader, (ii) att blåmeskomplexet har en kontinental härkomst och att Kanarieöarna koloniserades tre gånger, (iii) att samtliga öpopulationer har låg genetisk variation, vilket indikerar låga långfristiga effektiva populationsstorlekar och (iv) att populationer på La Palma och i Libyen utgör kvarlevorna av en population med nordafrikanskt ursprung. Vidare visade demografiska rekonstruktioner (v) att Kanarieöarna, i enlighet med den traditionella uppfattningen, har målpopulationer, vilka inte har utgjort någon källa till återkolonisation av det afrikanska fastlandet. Vår studie påvisar vikten av att samtliga taxa representeras, samt av en studiedesign som omfattar ett stort antal markörer, för att erhålla robusta fylogeografiska inferenser.
Introduktion
Isolerade öar är ofta bebodda av växter och djur som inte återfinns någon annanstans. Kända exempel omfattar karibiska anolisödlor (Losos 2011), Galapagosfinkar (Grant & Grant 2011), hawaiianska spindlar (Gillespie et al.1994) och Lord Howe-palmer (Savolainen et al. 2006). Man kan fråga sig hur dessa öar blev koloniserade: vilka var anfäderna på fastlandet, och när, varför samt hur uppstod diversiteten? Öbiota har inspirerat generationer av forskare, och spelat en central roll för utvecklingen av modern evolutions- och ekologiteori (Warren et al. 2015). Än i dag lockas evolutionsbiologer till isolerade öar, och efter inträdandet i genomikens era finns det en tilltagande optimism för att kartlägga mönster och processer kring kolonisation, diversifiering på öar, samt förhållanden till fastlandssläktingar.
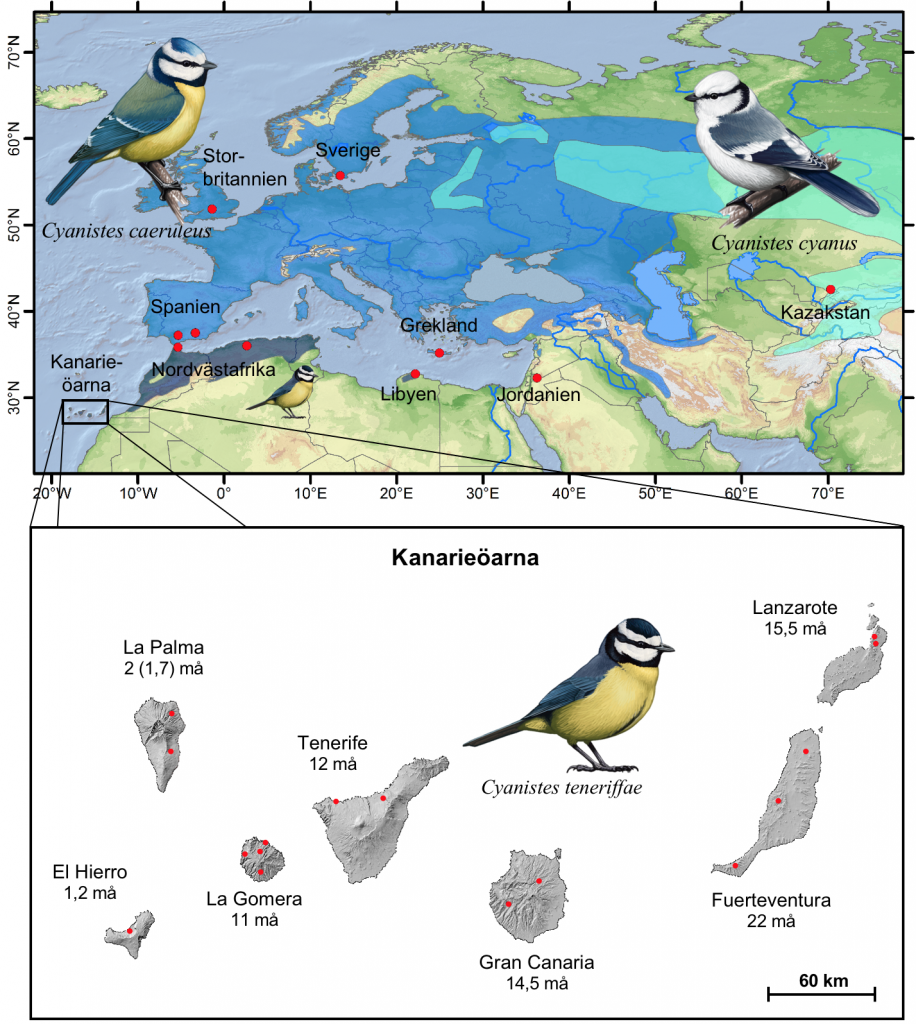 Figur 1. Provtagningsplatser (röda markeringar) och utbredningen av afrikansk blåmes Cyanistes teneriffae (mörk koboltblå), eurasisk blåmes C. caeruleus (mellanblå) samt azurmes C. cyanus (ljus cyanblå). Den infällda kartan visar Kanarieöarna och deras uppskattade geologiska ålder. Illustrationer av Martí Franch. Utbredningsdata från BirdLife International och NatureServe (2014), med modifikationer för den västliga delen av azurmesutbredningen enligt Gosler & Clement (2007); geologiska åldrar (i miljoner år; må) från Carracedo & Day (2002) och alternativ ålder för La Palma inom parentes, efter Guillou et al. (2001).
Figur 1. Provtagningsplatser (röda markeringar) och utbredningen av afrikansk blåmes Cyanistes teneriffae (mörk koboltblå), eurasisk blåmes C. caeruleus (mellanblå) samt azurmes C. cyanus (ljus cyanblå). Den infällda kartan visar Kanarieöarna och deras uppskattade geologiska ålder. Illustrationer av Martí Franch. Utbredningsdata från BirdLife International och NatureServe (2014), med modifikationer för den västliga delen av azurmesutbredningen enligt Gosler & Clement (2007); geologiska åldrar (i miljoner år; må) från Carracedo & Day (2002) och alternativ ålder för La Palma inom parentes, efter Guillou et al. (2001).
Öpopulationer är ofta betraktade som underordnade dem på fastlandet, och antas vara ett resultat från kolonisationen av de senare. Det finns omfattande exempel på detta, såsom kolonisationen av Darwins finkar på Galapagosöarna (Grant & Grant 2011), glasögonfåglar i Guineabukten (Melo et al. 2011) samt finkliknande vävare på Mauritius och Réunion (Warren et al. 2012). Den allmänna synen på öar som kolonisationsmål har emellertid förändrats på senare tid, och det finns numera exempel där öpopulationer har utgjort källan för omvänd fastlandskolonisation (Bellemain & Ricklefs 2008). Till exempel omfattar spridningen av monarker (familjen Monarchidae) från den papuanska arkipelagen, kolonisationer från arkipelagen till Sydostasien (Filardi & Moyle 2005), och liknande exempel kommer från kråkfåglar och släktingar (superfamiljen Corvoidea; Jønsson et al. 2011) och andra taxa (Balka et al. 2009; Jønsson et al. 2010). De nyligen dokumenterade exemplen av återkolonisation, från öar till fastland, har lett till en omvärdering av öars evolutionära och biogeografiska roller i en bredare geografisk kontext.
Kanarieöarna, som ligger ungefär 100 km utanför den nordvästafrikanska kusten, är en del av Medelhavsområdets högaktivitetszon för biologisk mångfald (Medail & Quezel 1999), med många endemiska växt- och djurarter, däribland fem endemiska fågelarter och flertalet underarter (Juan et al. 2000; Illera et al. 2012). Öarna sträcker sig över 500 km från öst till väst i en gradient av sjunkande ålder från 23 miljoner år till 1 miljon år (Fig. 1; Carracedo & Day 2002). Bland fågelpopulationerna i kanarieösamhället är den afrikanska blåmesen Cyanistesteneriffaedet mest utbredda och mångfaldiga taxonet, med fem erkända underarter (enligt the den internationella ornitologiska unionen, IOC; Gill & Donsker 2014): C. t. palmensispå La Palma, C. t. ombriosuspå El Hierro, C. t. teneriffaepå La Gomera och Tenerife, C. t. hedwigae på Gran Canaria, samt C. t. degener på Fuerteventura och Lanzarote. Därtill förekommer C. t. ultramarinusi Marocko, Algeriet, Tunisien och Pantelleria (en liten italiensk ö i Siciliensundet), och en isolerad population, C. t. cyrenaicae, förekommer i nordöstra Libyen. Utanför den afrokanariska regionen finns det ytterligare två arter i släktet: eurasisk blåmes C. caeruleus, förekommande i Europa och västra Asien med nio erkända underarter, och azurmes C. cyanus, som påträffas i Asien från Stilla havskusten ända till östra Europa, med åtta underarter (Gill & Donsker 2014). För ytterligare detaljer om utbredningen av taxa, se Fig. 1
De afrikanska blåmespopulationerna på Kanarieöarna har rönt stort intresse för deras avvikande fjäderdräkt (Martin 1991; Cramp & Perrins 1993), strukturella morfologi och ekologi (Partridge & Pringmill 1977; Grant 1979; Carrascal et al. 1994) samt sång (Schottler 1993, 1995). Eftersom de olika taxa är allopatriska blir det svårt att direkt testa tendenser till hybridisering, men sånguppspelningsexperiment utförda av Schottler (1995) visade att sångdifferentieringen mellan somliga ötaxa har fortskridit så mycket att sång från andra populationer inte ger någon respons, och Sangster (2006) menade att de förmodligen bäst skulle tilldelas artstatus.
Även om den populationsgenetiska strukturen hos blåmesarna på Kanarieöarna är välstuderad, är den afrokanariska blåmesens fylogeografiska historia ytterst komplex, och har varit svår att analysera, mot bakgrund av att varje enskild studie har föreslagit avsevärt till måttligt skilda scenarier (Salzburger et al. 2002; Kvist et al. 2005; Dietzen et al. 2008; Illera et al. 2011; Päckert et al. 2013; Hansson et al. 2014; Gohli et al. 2015). Således förblir antalet kolonisationstillfällen (1–3) omtvistat, liksom kolonisationsbanan inom och mellan Kanarieöarna och fastlandet (stegvis kolonisation i västlig eller östligriktning, eller från kärnan utåt), samt ursprunget till den genetiskt avvikande populationen på La Palma (från Europa eller Afrika). Den senare har blivit grupperad med andra afrokanariska populationer (Kvist et al. 2005; Päckert et al. 2013; Gohli et al. 2015), men även med eurasiska populationer tack vare likhet i sekvens och homologa förluster av DNA-baser (Dietzen et al. 2008).
Vidare förblir Kanarieöarnas betydelse kontroversiell som kolonisationsmål från fastlandskällor eller som källa för fastlandskolonisation. Åtminstone till en viss utsträckning kan denna osäkerhet förklaras med förekomsten av kraftiga flaskhalsar och ofullständig härkomstssortering i Kanarieökladen, vilket antyder att olika loci ofta har olika evolutionär historia. Dessvärre har de flesta studier av den afrokanariska blåmesen varit baserade på en mitokondriell markör (Salzburger et al. 2002; Kvist et al. 2005; Dietzen et al. 2008), en eller ett fåtal ytterligare kärnmarkörer (Illera et al. 2011; Päckert et al. 2013), och/eller lidit av ofullständig eller inadekvat provtagning av relevanta taxa, i synnerhet den libyska underarten C. t. cyrenaicae(Salzburger et al. 2002; Kvist et al. 2005; Dietzen et al. 2008; Illera et al. 2011; Hansson et al. 2014; Gohli et al. 2015).
Den senaste utvecklingen inom sekvenseringsteknologi och bioinformatik möjliggör avsevärt större genomiska dataset och utlovar nya insikter om omstridda biogeografiska och evolutionära koncept (Davey et al. 2011; Ekblom & Galindo 2011). Dessa teknologiska fördelar kräver även nya analystyper, och enkelgensfylogenier är ersatta med fylogenetiska analyser av sammanbundna sekvensmatriser, eller koalescensbaserade artträdsslutsatser (se t.ex. Patel et al. 2013). I en nyligen publicerad studie använde Gohli et al. (2015) parallellsekvensering (s.k. ”next-generation sequencing”), men de hade endast tillgång till degraderade museiprover av den libyska underarten, vilket gav upphov till en fylogeni med lågt stöd för de huvudsakliga kladerna. Dessutom saknar deras huvudsakliga koalescensbaserade artträd och demografiska analys (exklusive den libyska populationen) starkt statistiskt stöd (dvs. artträdsanalyserna förefaller konvergera dåligt och den effektiva provtagningen är i de flesta fall för låg), vilket gör de publicerade evolutionära tolkningarna osäkra.
I den här studien undersöker vi förhållandet mellan afrokanariska blåmestaxa från fastlandet och motsvarande från öarna, samt deras förhållande till de eurasiska Cyanistes-taxa, med hjälp av en kombination av traditionell Sangersekvensering av flera gener på populationsnivå och parallellsekvensering av enskilda populationsrepresentanter, analyserade i koalescensbaserade, fylogenetiska och strukturindelningsbaserade ramverk. Med den kombinerade styrkan av dessa olika ramverk ämnar vi förstå den afrokanariska blåmesens evolutionshistoria, dess inbördes förhållande samt dess förhållande till eurasiska släktingar. Särskilt riktar vi in oss på antalet kolonisationstillfällen till Kanarieöarna, kolonisationsbanan/-orna, och vi uppskattar tidpunkterna för dessa tillfällen. Slutligen avser vi att stödja eller ifrågasätta rollen för Kanarieöarnas blåmesar som grundare av den utbredda populationen på det afrikanska fastlandet.
Material och metod
Fjäder- eller blodprover samlades in från slöjnätsfångade vilda fåglar i afrikanska blåmespopulationer på samtliga sju kanarieöar, i nordvästra Afrika (Ceuta, fortsättningsvis refererat till som Marocko, och Algeriet), i Libyen samt tre eurasiska blåmespopulationer i Spanien, Storbritannien och Sverige (Tabell S1, Stödinformation, Fig. 1). Från varje population togs prover från 4–5 individer för Sangersekvensering, liksom 1–2 individer från Grekland och Jordanien, samt azurmes och talgoxe Parus major. Från samtliga populationer var en enskild representant utvald för RAD-sekvensering (restriktionspunktsassocierad DNA-sekvensering), tillsammans med ytterligare utgruppspopulationer såsom Periparus spp. och Poecile spp. (för detaljer, se Tabell S1, Stödinformation). Blodprover förvarades i etanol eller SET-buffert. DNA extraherades genom standard fenol-kloroformprotokoll för blod (Sambrook & Russel 2001) eller standard sodiumacetatmetod för fjädrar. DNA som extraherades från fjäderprover var helgenomsamplifierade genom REPLI-g-satsen (Qiagen) enligt tillverkarens instruktioner.
Sangersekvensering av populationsprover
Amplifiering och Sangersekvensering. Vi amplifierade DNA genom Qiagen Multiplex PCR Kit i reaktioner om 10 µl, för två mitokondriella och 18 kärnmarkörer. För detaljer om prajmrar och PCR-profiler, se Tabell S2, Stödinformation. PCR-produkter precipiterades (NH4Ac och etanol) och löstes därefter i 10–40 µl vatten, beroende på amplifieringsresultat, av vilka 2 µl användes för 10 µl sekvenseringsreaktion (BigDye sekvenseringssats; Applied Biosystems; Tabell S2, Stödinformation) i 35 PCR-cykler på 96 °C i 10 s, 47 °C i 5 s och 60 °C i 4 min. Produkterna avlästes på en ABI Prism 3100 kapillärsekvenserare (Applied Biosystems).
Sekvensredigering, sekvensinpassning och haplotypinferens. Sangersekvenserna redigerades med Geneious 6.1 (Biomatters). Vi trimmade sekvenserna och letade efter dubbla toppar i elektroferogrammen med hjälp av insticksmodulen för att hitta heterozygoter. Sekvensfragment med oklarheter på grund av låg kvalitet kodades som saknad data. Vi inpassade sekvenserna med hjälp av insticksmodulen MAFFT (Katoh & Standley 2013) för sekvensinpassning. För att erhålla fasad data från Sangersekvenserigsdatasetet infererade vi haplotyper med PHASE v. 2.1-algoritmen (Stephens et al. 201; Stephens & Donnelly 2003), tillgänglig i DNASP v. 5.10.01 (Librado & Rozas 2009). För vidare detaljer om haplotypinferens, se Stödinformation.
Val av substitutionsmodell och implementering av en molekylär klocka. Substitutionsmodeller utvärderades med JMODELTEST v. 2.1.4 (Guindon & Gascuel 2003); Darriba et al. 2012), genom val från 88 tillgängliga modeller med heterogenitet enligt fyra gammakategorier samt en del oföränderliga positioner i beaktande. Modellurval genomfördes enligt Bayes Informationskriterium (BIK; Schwarz 1978). Vi använde inställningar för molekylklockan enligt följande: för cytokrom b, 0,0105 ± 0,0005 (95 % konfidensintervall) substitution/position/Ma (Weir & Schluter 2008); för kontrollområdet, den Cyanistesspecifika beräkningen 0,02 ± 0,01 substitution/position/Ma (Päckert et al. 2007); och för kärnmarkörer, 0,00135 ± 0,00045 substitution/position/Ma (Ellegren 2007). Gränserna för 95 % konfidensintervall användes som gränser för ingångsvärden med en enhetlig fördelning och molekylklockan var, såvida inget annat uppges, modellerad med strikta ingångsvärden.
Trädanalyser av koalescenta arter. *BEAST gör inferens av ett artträd parallellt med markörspecifik trädrekonstruktion (Heled & Drummond 2010). Indata förbereddes i BEAUti, del av BEAST v. 2.0.2-sviten (Bouckaert et al. 2014) tillsammans med *BEAST. Estimates av JMODELTEST av transitions-/transversionsförhållande, andelen oföränderliga positioner och gammaformsparameter implementerades, medan basfrekvenser lämnades för empirisk beräkning (utom K80-modeller). Klockmodeller var sammankopplade för samtliga kärnmarkörer, och trädmodeller kopplades ihop för de två mitokondriella markörerna, medan andra träd- och substitutionsmodeller lämnades okopplade. Vi ställde in ingångsvärdena för artträd att följa Yuleprocessen och populationsstorleksmodellen att vara styckevis linjär med konstant rot.
I BEAST genomförde vi två självständiga MCMC-körningar med en kedjelängd på >850M, med insamling per 100K. Utdata undersöktes med TRACER v. 1.5 (Rambaut & Drummond 2007), för att säkerställa att likhetsvärdet var fast och att den effektiva provstorleken (ESS) var adekvat, med inbränningeninställd på 10 %.
Ytterligare *BEAST-analys utan de två mitokondriella markörerna – och således endast med kärnmarkörer – genomfördes enligt ovanstående.
Analyser av genetisk variation och demografisk rekonstruktion. Populationsspecifik nukleotiddiversitet, haplotypdiversitet och θ beräknades med hjälp av DNASP v. 5.10.01 (Librado & Rozas 2009).
Vi rekonstruerade historisk demografi genom utökad Bayesiansk siluettdiagramsanalys (Heled & Drummond 2008) i BEAST v. 1.8.0 (Drummond et al. 2012). Analyserna baserades på variabla loci (se Tabell S3, Supportinformation) och tillämpade klockor och ingångsvärden för träd enligt specifikationerna ovan för *BEAST-analyser, med vissa modifikationer för operatörsbelastning. Analyser gjordes för 500M generationer, med insamling per 40K generationer. Resultat fastställdes med hjälp av GGPLOT2 v. 1.0-paket (Wickham 2009) i R v. 3.0.3 (R Core Team 2013).
RAD-sekvensering av individuella representanter
Vi analyserade RAD-markörer spridda över hela genomet, för enskilda representanter per population, i enlighet med uppfattningen att optimering av resolution och precision i fylogenetisk inferens bäst uppnås genom att öka antalet markörer, snarare än antalet individer (Corl & Ellegren 2013).
Bibliotekskonstruktion samt sekvensering. Vi förberedde ändparade RAD-bibliotek för en individ per population samt ytterligare utgrupper, för släktena Periparusoch Poecile, enligt protokollen av Baird et al. (2008) och Etter et al. (2011) med modifieringar enligt Stödinformation. Sex prover visade lägre sekvenseringstäckning (i synnerhet C. t. degenerfrån Lanzarote och P. major) och inkluderades därför i ett efterföljande bibliotek. För dessa prover genomfördes upprepad underbindning med adapter P1 av DNA från det första biblioteket, vilken redan var enzymatiskt nedbruten och underbunden med P1-adapter, samt inkluderades i återstoden av bibliotekskonstruktionen. För RAD-sekvensering och datafiltreringsresultat, se Tabell S4, Stödinformation.
Bioinformatisk processkedja. De råa avläsningarna av ändparat DNA filtrerades först med hjälp av komponenterna process_radtagsoch clone_filteri STACKS v. 1.17 (Catchen et al. 2013). Avläsningar av prover, vilka hade inkluderats i två bibliotek, slogs sedan samman per prov. Vi kartlade återstoden av avläsningarna till zebrafinksgenomet Taeniopygia guttata(version taeGut3.2.4, nedladdat från UCSC:s genomwebbplats http://genome.ucsc.edu/) med BOWTIE V 2.2.2 (Langmead & Salzberg 2012). Givet den interspecifika kartläggningen, fastställde vi maximi- och minimistraff för felmatchning (–mp) till 2 respektive 1. Vidare godtog vi enbart ändparade avläsningar i den förväntade relativa riktningen inom 150–1000 baspar (bp) från varandra (–no-discordant,–no-mixed). Från de resulterande SAM-filerna avlägsnade vi avläsning 2 och använde avläsning 1 som indata till STACKS, följande ref_map-spåret för fylogenetiska och koalescensanalyser. Vi använde standardparametrar utom för att ställa in en högre minimisekvenseringstäckning för att kalla ett locus i ett givet prov (m= 4). Provet påC. t. degenerfrån Lanzarote, liksom P. major-provet var av substantiellt lägre täckning, och ytterligare databehandling genomfördes parallellt för ett dataset i vilket de två proverna behölls, och ett i vilket de två proverna togs bort.
Vi filtrerade utdatan så att det endast innehöll loci (i) i vilka samtliga prover tilldelats 1–2 alleler; (ii) vilka innehöll 1–10 singelnukleotida polymorfismer (SNP:er); (iii) vilka omfattade ≤ 12 alleler. Syftet med dessa filter var att eliminera eventuella loci i vilka individer tilldelats mer än två alleler och/eller vilka var avsevärt växlande (avbrytningsvärden var emellertid godtyckligt tilldelade efter granskning av data). Genom att göra så kan vi ha alternerat felaktig uteslutning av kraftigt växlande loci för att inte inkludera felaktigt sammansatt loci.
Utdatan från STACKS konverterades till bialleliska sekvenser för samtliga prover och loci, genom skräddarsydda skript, och sammanlänkades baserat på prov (med slumpvis allelparning) till långa supermatriser. Avslutningsvis definierade vi ett dataset i vilketall taxa inkluderades, och ingen saknad data för något locus accepterades: supermatris STRICT, innefattande 11 426 loci med sammanlagd längd på 994 062 bp. I ett annat dataset avlägsnade vi två prover med lägsta täckning och tillät upp till 48 % saknad data bland de återstående proverna för varje enskilt locus: supermatris RELAXED, innefattande 37 292 loci med en sammanlagd längd på 3 244 404 bp.
Fylogenomiska analyser. Supermatriser STRICT och RELAXED analyserades i RAXML v8.0.26 (Stamatakis 2014), genom GTRGAMMA-substitutionsmodellen (allmän tidsreversibel modell med utrymme för heterogen mutationstakt, enligt en gammafördelning). Det bästa trädet för maximal sannolikhet (maximum likelihood; ML) från 1 000 sökningar valdes ut, och 1 000 s.k. bootstrap-upprepningar för supermatrisen STRICT, och 200 upprepningar för supermatrisen RELAXED, genomfördes. Konvergens verifierades med hjälp av ett bootstrap-test posteriori med autoMRE-funktionen (Pattengale et al. 2009).
Tabell 1. Uppskattade medelåldrar i miljoner år (95 % högsta posterioridensitet) för tre olika kalibreringar och fyra subset av 14-Kbp genomisk RAD-sekvensdata; tre oberoende autosomala (här presenterade med medelvärde) och en från Z-kromosomen. För kalibreringar och taxonset, se Tabell S5, Stödinformation).
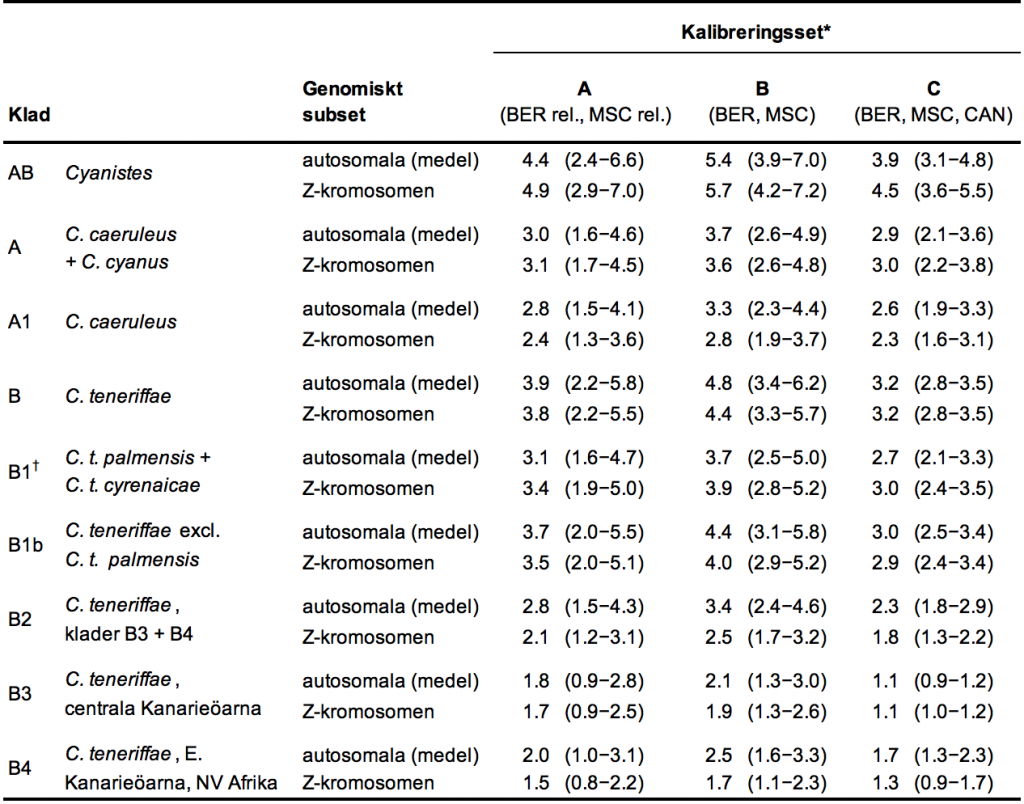 rel., relaxerad
rel., relaxerad
*Kalibreringspunkter gällde som följer: BER, Beringia Nearctic/Holarctic delat inom Pœcile för 5,32–5,33 miljoner år eller relaxerad för 0–5,96 miljoner år; CAN, Kanarieövulkanisk ålder för El Hierro 0–1,2 miljoner år och La Palma 0–3,5 miljoner år.
†C. t. palmensis (La Palma) och C. t. cyrenaicae (Libyen) specificerades som en monofyletisk klad enligt trädanalyser (se text).
Klusteranalyser. En multidimensionell skalanalys av det ofiltrerade SNP-datasetet på 3 670 SNP:er genomfördes i PLINK v. 1.9beta2c (Purcell & Chang 2014) för att upptäcka likheter och skillnader bland och inom de studerade populationerna.
Datering. Av databegränsningsskäl valde vi, från supermatris STRICT, subset omfattande 14 Kbp (160 loci) vardera, vilka vi använde för fylogenomisk datering. Vi valde slumpvis ut tre subset av autosomala loci genom funktionen sample {base} i R (R Core Team 2013) och gjorde ett subset av dessa loci som hade knutits till könskromosomen Z.
Ingångsvärdena för kalibrering placerades på delningen mellan nearktiska och holarktiska taxa av Poecile, separerade före eller vid den sista öppningen av Berings sund (för 5,5–5,4 miljoner år sedan enligt Marincovich & Gladenkov (1999) och Gladenkov et al. (2002)) och delningen mellan nordafrikanska och europeiska svartmespopulationerPeriparus ater, överensstämmande med salthaltskrisen i sena miocen (i sin helhet omfattande 5,96–5,32 miljoner år sedan enligt Krijgsman et al. (1999)). Till följd av argument från Päckert et al(2011) för att det faunala utbytet mellan det holarktiska och det nearktiska troligen inträffade före den sista öppningen av Berings sund, och jämförbart med Illera et al. (2011) och Päckert et al. (2013), använde vi en mer relaxerad kalibrering (A). Ingångsvärdena för den nearktisk-holarktiska delningen följde en enhetlig fördelning från 14–4,8 miljoner år sedan och en enhetlig fördelning från 5,96–0 miljoner år sedan applicerades för salthaltskrisen i sena miocen. Vi använde även ett antal alternativa kalibreringar, inklusive mer konservativa inställningar, för ovanstående kalibreringspunkter, till exempel ställa in ingångsvärden för delningen mellan nordafrikanska och europeiska svartmespopulationer för Medelhavets uttorkningsperiod (kalibrering B, D–E; Tabell 1, Tabell S5, Stödinformation).
Två ytterligare restriktioner enligt öars geologiska åldrar lades slutligen till (kalibrering C; Tabell 1 och S5, Stödinformation) i överensstämmelse med Päckert et al. (2013): Ingångsvärdena för La Palma (klad B eller B1), följde en enhetlig fördelning från 3,5–0 miljoner år sedan och El Hierro (klad B3) från 1,2–0 miljoner år sedan (till följd av Fernández-Palacios et al. 2011).
Samtliga dateringar genomfördes både för det fullständiga taxonsetet och för ett partiellt taxonset, från vilket proverna från Lanzarote och Teneriffa avlägsnades, med anledning av kraftigt högre inomindividuell genetisk variation än andra prover.
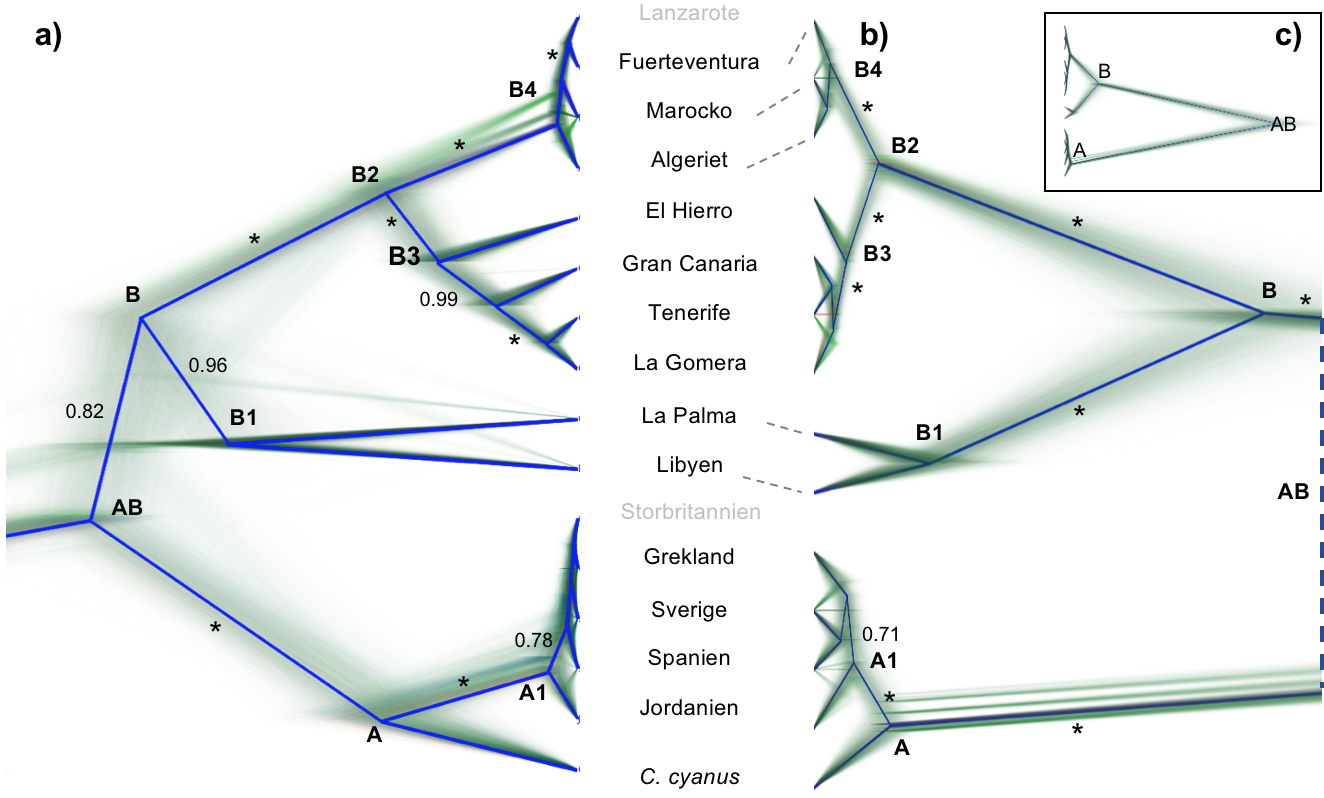
Figur 2. Koalescensbaserade artträd av blåmes Cyanistesspp. Träd är ritade i DensiTree med individuella träd representerade av mycket tunna linjer, som bildar träddensitet, och huvudtopologi accentuerat med en distinkt linje. Efterföljande sannolikhet högre än 0,6 är fastställda längs med grenarna (*=1,0). Artträden är baserade på (a) populationsprover från två mitokondriella och 18 kärnmarkörer sekvenserade enligt Sangermetoden, beräknade med *BEAST och (b, c) enkelnukleotida polymorfismdata (SNP) från restriktionspunktsassocierad DNA-sekvensering (RAD) av enskilda individer per härkomst, beräknat med hjälp av SNAPP. Notera att de artspecifika grenarna är systematiskt förkortade i det RAD-baserade trädet, på grund av metodologi (se text); därför är den avslutande delen av det RAD-baserade trädet inzoomat (b), medan den fullständiga topologin visas i en infälld panel (c). Kladnamn refereras till i texten. Populationer avC. teneriffae och C. caeruleusrefereras till genom deras geografiska ursprung, och beteckningar är inpassade med ändarna av sekvensbaserade träd (a). Det RAD-baserade trädet (b) inkluderar inte prover från Lanzarote eller Storbritannien (gråmarkerad), och streckade linjer indikerar inpassning med populationsbeteckningar när dessa inte är inpassade.
ML-trädet från supermatris STRICT (beskuret för det partiella taxonsetet) transformerades i R-paket APE v. 3.1.4 (Paradis et al. 2004) för att passa kalibreringsrestriktionerna i enheter om miljoner år, och dessa träd användes som startträd i dateringen av analyser genomförda i BEAST v. 2.1.3 (Bouckaert et al. 2014). För att tillgodose den stora inkongruensen mellan ML och koalescensartträden, utfördes upprepningar av samtliga dateringskörningar, med startträd i vilka populationerna av från Palma och Libyen gjordes gällande som en monofyletisk klad (jfr klad B1, Fig. 2).
De sammanlänkade sekvenserna var inte partitionerade, och en okorrelerad, lognormal, flexibel klocka implementerades (Drummond et al. 2006). Vi använde en Yule-artbildningsmodell, och ställde in ingångsvärdena enligt en diffus gammafördelning på födelsetakten. Stora klader begränsades som monofyletiska i enlighet med startträdets topologi, vilket rotades enligt de intergeneriska förhållandena i Johansson et al. (2013). Vi körde MCMC-kedjan 100M generationer, med insamling per 10K generationer, och undersökte resultaten med TRACER v. 1.5 (Rambaut & Drummond 2007), efter vilket 10 % av träden förkastades som inbrända, och maximal trovärdighetskladträd beräknades i TREEANNOTATOR (Bouckaert et al. 2014). För 3 av 80 analyser, var stationaritet inte uppnådd inom 10M generationer, och effektiv provstorlek var inte >>200, och dessa kördes ytterligare 10–25M generationer, varefter inbränning avfärdades så att det slutliga antalet eller analyserade träd motsvarade samtliga andra dateringsanalyser.
Koalescenta artträdsanalyser av SNP:er. Med hjälp av VCFTOOLS v. 0.1.12a (Danecek et al. 2011), extraherade vi ingruppen enbart från supermatris STRICT och valde effektivt ickelänkade SNP:er genom att tillåta ett maximum på en SNP per RAD-locus. Detta ofiltrerade SNP-set innehöll 3670 utspridda SNP:er. I beräkningsmässigt syfte filtrerades autapomorfier bort från SNP-setet genom att kräva ≥3 förekomster av en alternativ allel, vilket resulterade i bevarande av 3421 SNP:er. Det filtrerade datasetet importerades till BEAUTI (Bouckaert et al. 2014), där ingångsvärdena för framåt- (u) och bakåtmutationsgraden (v) initialt var inställda på 1/(2*allelfrekvens) för referens (u) och alternativ (v). Emellertid skapade preliminära körningar förbättrad effektivitet när u- och v-parametrar meduppskattades under körningarna, varför meduppskattning implementerades i slutgiltiga körningar. Återstående parametrar lämnades på standardvärden, exempelvis artdivergensgraden λ = 0,00765, och θ definierad av ingångsvärden som följde en γ-fördelning med formparameter α = 11,750 och skalparameter β = 109,73. Vi utförde två oberoende körningar för ≥8,4M generationer, med insamling per 1K generationer med hjälp av insticksmodulen SNAPP v. 1.1.16 (Bryant et al. 2012) i BEAST v. 2.1.3 (Bouckaert et al. 2014). Utdata undersöktes med TRACER v. 1.5 (Rambaut & Drummond 2007), och de konvergerande körningarna kombinerades efter att ha uteslutit 10 % som inbrända. Effektiv provstorlek för ingångsvärden, utfallsvärden, sannolikhet och trädhöjd var >>200 (medan beräkningar av θ för forntida populationer, nådde – som förväntat – inte tillräckliga effektiva provstorlekar baserade på en individ per population).
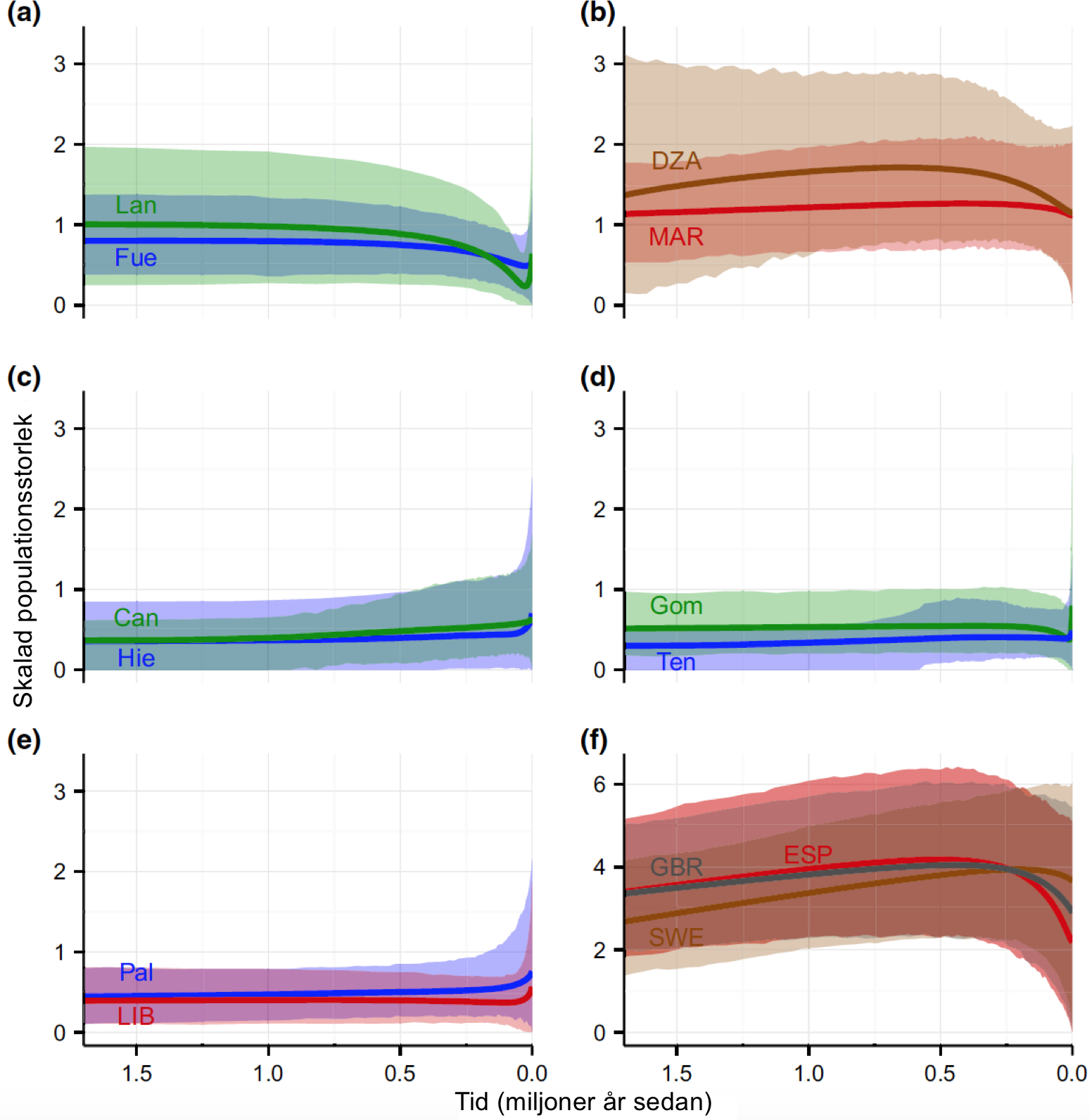
Figur 3. Demografisk rekonstruktion av populationer av afrikansk blåmes Cyanistes teneriffae (a–e) och eurasisk blåmes C. caeruleus (f) med hjälp av utbyggt Bayesianska siluettdiagram baserade på Sangersekvensdata från flera markörer. Heldragna linjer representerar beräknad skalenlig medelpopulationsstorlek (effektiv populationsstorlek X generationstid i miljoner år), och skuggade områden de 95 % högsta posterioritäthet. Populationer är märkta med förkortningar och ötaxa är färgade i blått/grönt, medan fastlandspopulationer är färgade i rött/brunt/grått. (a) Östra Kanarieöarna: Fuerteventura (Fue; blått) och Lanzarote (Lan; grönt), (b) nordvästra Afrika: Marocko (MAR; rött) och Algeriet (DZA; brunt), (c) centrala Kanarieöarna: Gran Canaria (Can; grönt) och El Hierro (Hie; blått), (d) östra Kanarieöarna: La Gomera (Gom; grönt) och Teneriffa (Ten; blått), (e) La Palma (Pal; blått) och Libyen (LIB; rött), (f) europeiska populationer: Spanien (ESP; rött), Storbritannien (GBR; grått) och Sverige (SWE; brunt). Notera att den sista panelen (f) har en annorlunda skala på y-axeln. Alternativa versioner av den här figuren, utzoomat för att täcka den fullständiga rekonstruktionen, respektive inzoomat på det mest aktuella perioden, är tillgänglig som Fig. S3 och S4, Stödinformation.
Resultat
Sangersekvensering av populationsprover
Sangersekvenseringsdatan inkluderade 4–5 individer per population och täckte sammanlagt 12 498 bp fördelat över 18 kärn- och två mitokondriella markörer. För resultaten av urvalet för substitutionsmodell för Sangersekvenseringsmarkörer, se Tabell S6, Stödinformation.
Koalescensartträdsanalyser. De två sekvensbaserade artträdskörningarna baserade på mitokondriella och kärnmarkörer, i *BEAST konvergerade, och resultatdata kombinerades, vilket gav till följd ett slutgiltigt set om 15 656 träd. De skapade artträden reflekterar en eurasisk klad (klad A) med fullständigt stöd (efterföljande sannolikhet (ES) = 1,0) [Vill man behålla PP som förkortning, kanske posteriör probabilitet är att föredra, även om det är rena fikonspråket. Språkrådet rekommenderar dock efterföljande sannolikhet.] med eurasisk blåmes (klad A1) och azurmes och en fullständigt stödd afrokanarisk klad (klad B), till vilken populationerna från Libyen och La Palma är tilldelade med ES = 0,82 (Fig. 2a). Inom den afrokanariska kladen, rymmer en basal gren den libyska och La Palma-populationerna, grupperade som systertaxa med ES = 0,96 (klad B1). Återstående taxa divergerar i en olöst och ytlig grupp från Nordvästafrika och östra Kanarieöarna (dvs. Fuerteventura och Lanzarote; klad B4) och en grupp innehållande centrala Kanarieöarnas populationer (klad B3), båda med fullständigt stöd. I det sistnämnda, är El Hierro basal, följt i förgreningsordning av Gran Canaria och sedan La Gomera/Teneriffa (ES ≥0,99 för alla noder; Fig. 2a).
De markörspecifika träden varierar i topologi, men de snabbutvecklande mitokondriella markörerna återfick den allmänna artträdstopologin inklusive alla större klader (se t.ex. träd för kontrollregionen, Fig. S1, Stödinformation). I dessa träd blev samtliga populationer inom kladerna B1 och B3 ömsesidigt monofyletiska, medan det finns parafyli med hänsyn till populationer inom kladerna A1 och B4 (Fig. S1, Stödinformation).
Körningen utan mitokondriella markörer, resulterade i ett artträd med fullständigt stödda klader B3 och B4 (Fig. S2, Stödinformation), identiska i topologi med artträden med mitokondriella markörer (Fig. 2a). Lösning på ett djupare evolutionärt plan är emellertid sämre: den basala delningen av Cyanistesskiljer populationen från La Palma från alla andra populationer, följt av förgrenande europeiska populationer (klad A), därefter den libyska populationen och slutligen Kanarieökladerna B3 och B4 (Fig. S2, Stödinformation). Den efterföljande sannolikheten för noderna innefattande samtliga populationer förutom La Palma är 0,70, och för noden innefattande kladerna B3, B4 och Libyen 0,74. Vidare återfinns C. cyanus, i klad A, bland C. caeruleus-populationer.
Genetisk variation och demografisk historia. Den genetiska diversiteten är generellt låg bland populationerna på Kanarieöarna, i synnerhet i El Hierro- och Lanzarotepopulationerna (Tabell S3, S7 och S8, Stödinformation). Bland afrikanska fastlandspopulationer visar den libyska, i paritet med de från Lanzarote, låg genetisk variation. Den marockanska och, särskilt, den algeriska populationen visar medelhög diversitet och θ-värden vilka är högre än de för Kanarieöarna och Libyen, men lägre än de för europeiska populationer, vilket, i sin tur, visar relativt hög diversitet och θ-värden (Tabell S3, S7 och S8, Stödinformation).
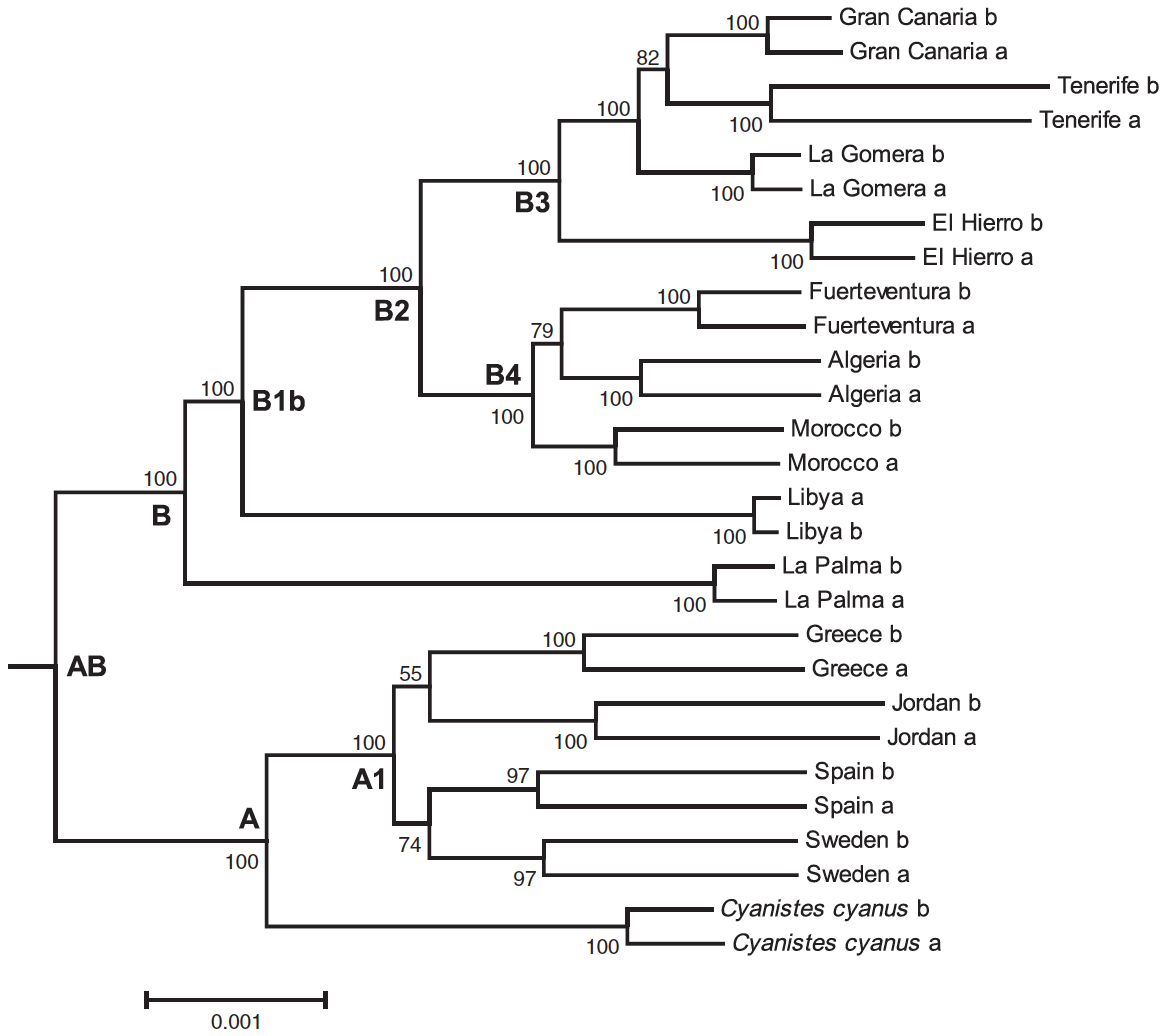
Figur 4. Fylogenomiskt maxsannolikhetsträd uträknat i RAXML från supermatris RELAXED, innefattande 37 292 restriktionspunktsassocierade DNA-sekvenser (RAD-sekvenser) (3,244 Mbp) från samtliga individer förutom lågtäckningsprover (dvs. Lanzarote och P. major) med tillåtelse för upp till 48 % saknad data för varje locus. I sekvensnamnen betecknar “a” och “b” de två “allelerna” producerade genom slumpvist kombinerade alleler från varje enskilt RAD-locus. Bootstrap-stöd >50 visas vid noder; klader refereras till i texten; utgrupper som inte visas.
Rekonstruktionen av demografisk historia med Bayesiansk siluettdiagram visar generellt högre medelstorlek för fastlandspopulationerna, förutom Libyen, än för öpopulationerna, även om konfidensintervallen är stora (Fig. 3, S3 och S4, Stödinformation). De flesta öpopulationer visar tecken på en nyligen inträffad expansion, och i populationerna från Lanzarote, La Gomera och till viss del Fuerteventura, föregås det av flaskhalsar (Fig. 3a, c–e och S4a, c–e, Stödinformation). Rakt motsatt visar fastlandspopulationerna från Nordvästafrika, såväl som de europeiska populationerna, inga tecken på flaskhalsar, och trenderna för populationsstorleken sträcker sig över längre tid (Fig. 3b, f och S3b, f, Stödinformation).
RAD-sekvensering av enskilda representanter
Enstaka individer per population, tillsammans med ytterligare utgrupper, var subjekt till RAD-sekvensering, vilket resulterade i reducerad representationsgenomisk data (Tabell S4, Stödinformation). Olika datafiltrering skapade två huvudsakliga dataset av sammanlänkade RAD-loci; supermatris STRICT som inbegrep samtliga taxa och inte tillät någon saknad data (11 426 loci innehållande 994 Kbp) och supermatris RELAXED som exkluderade två lågtäckningsprover och tillät upp till 48 % saknad data (37 292 loci innehållande 3,24 Mbp; se Material och metod).
Fylogenomiska analyser av sammanlänkad sekvenseringsdata. De fylogenomiska träden, infererade av RAXML skiljer sig mycket lite mellan supermatris STRICT (Fig. S5, Stödinformation) och supermatris RELAXED (Fig. 4). Den övergripande topologin i de sammanlänkade träden är liknande de koalescenta artträden, med en eurasisk och en afrokanarisk klad. Det finns, emellertid, vissa olikheter: Populationerna på La Palma och i Libyen ar inte infererade som systertaxa vilka delat sig från en lång gren. I stället grenar La Palma av först, varefter Libyen [jag skiter i “shortly thereafter”] grenar av från stamfadern av de återstående afrokanariska populationerna (Fig. 4 och S5, Stödinformation). För supermatris STRICT (Fig. S5, Stödinformation), den centralkanariska kladen (klad B3) matchar artträdens topologi (cf. Fig. 2), medan El Hierro identifieras som basal för supermatris RELAXED, varefter La Gomera grenar av, och slutligen Gran Canaria/Teneriffa (det sistnämnda, emellertid inte med full bootstrap-stöd; Fig. 4).
I supermatris STRICT är klad B4 löst med Algerietbasal, följd av Marocko i greningssekvens, och Fuerteventura/Lanzarote (Fig. S5, Stödinformation), medan greningsskevensen för supermatris RELAXED blandar kontinentala och insulära populationer (Fig. 4). De flesta noder är stödda med högt bootstrapsvärde (Fig. 4 och S5, Stödinformation).
För RAD-sekvenseringsdata är genetisk variation inom arter (inom prover) för C. t. degenerfrån Lanzarote och C. t. teneriffae från Teneriffa anmärkningsvärt högre än för andra prover (Fig. S5, Stödinformation).
Klusteranalyser. Den mångdimensionella skalanalysen av det ofiltrerade SNP-datasetet producerade sex kluster, motsvarande populationsgrupper från Eurasien (klad A), centrala Kanarieöarna (klad B3, med Teneriffa tämligen avvikande), Nordvästafrika och östra Kanarieöarna (utom Lanzarote, som avviker från återstående klader B4), La Palma och Libyen (Fig. S6, Stödinformation).
Datering. Autosomal sekvensdatering baserad på relaxerade inställningar för två kalibreringspunkter (kalibrering A), den interkontinentala delningen mellan holarktisk och nearktisk Pœcile-arter samt den afrikanska/europeiska delningen inom Periparus aterföljd av salthaltskrisen i sena miocen, skapade de följande medelåldersberäkningar för partiell taxonset exklusive prover från Lanzarote och Teneriffa: Klad AB (samtliga Cyanistes) 4,4 miljoner år, klad B (C. teneriffae) 3,9 miljoner år, klad B1b (C. teneriffae utom La Palma) 3,7 miljoner år eller klad B1 (La Palma och Libyen) 3,1 miljoner år, klad B3 (centrala Kanarieöarna) 1,8 miljoner år, klad B4 (östra Kanarieöarna och Nordvästafrika) 2,0 miljoner år, samt klad A (C. caeruleusoch C. cyanus) 3,0 miljoner år (Tabell 1). De fullständiga resultaten från samtliga reproduktionsanalyser är tillgängliga i Tabell S5, Stödinformation.
Det fanns en del effekter av genomiskt subset: I förhållande till autosomalt subset 1 inträffade autosomalt subset 2 och 3 vid i genomsnitt 13 % respektive 6 % högre nodåldersuppskattning, medan Z-kromosomsubsetet producerade i genomsnitt 2 % högre nodåldrar, för det fullständiga taxonsetet. Dessa skillnader var mer uttalade för det partiella taxonsetet: I förhållande till autosomalt subset 1 inträffade autosomalt subset 2 och 3 vid i genomsnitt 28 % respektive 17 % högre nodåldersuppskattning, medan Z-kromosomsubsetet producerade i genomsnitt 7 % högre nodåldrar.
Effekten av att exkludera två prover, tack vare oväntat hög inomindividuell genetisk variation, hade en större effekt på de uppskattade nodåldrarna (Tabell S9, Stödinformation). Effektstorleken är oföränderlig över samtliga kalibreringar, utom den som införlivar restriktioner av Kanarieöarnas geologiska åldrar (A, B, D och E), men varierar märkbart mellan specifika noder. För nod A och A1 (eurasisk C. caeruleusoch C. cyanus) är effekten i genomsnitt 10 %; för noderna AB, B och B1/B1b 24 %; 43 % för nod B2; 55 % för nod B3; och 72 % för nod B4 (Tabell S9, Stödinformation).
Konfigurationen av ingångsvärdena för kalibrering påverkade dateringen (av det partiella taxonsetet) enligt följande: genom att ställa in kalibreringspunkten för salthaltskrisen i sena miocen att inkludera enbart perioden med faktisk uttorkning av Medelhavet (SKM strikt), snarare än att relaxera det föregående genom att endast ställa in en övre tidsgräns, ökar de uppskattade nodåldrarna enhetligt med i genomsnitt 22 % (Tabell S10, Stödinformation). Dock avviker skillnaden i uppskattade nodåldrar över noder endast med 2 % mellan relaxerade och strikta ingångsvärden för det transberingska faunautbytet (Tabell S10, Stödinformation). När strikta ingångsvärden för salthaltskrisen i sena miocen appliceras, uteblir effekten från att lägga till ingångsvärden för det transberingska faunautbytet (Tabell S10, Stödinformation). Att lägga till restriktioner i enlighet med de geologiska åldrarna av El Hierro och La Palma skapade uppskattade nodåldrar som är 21–31 % lägre, utom för kladen för centrala Kanarieöarna B3, vilken var 45 % lägre.
Slutligen hade upprätthållandet av klad B1 i enlighet med koalescensartträdstopologin ingen effekt på de uppskattade nodåldrarna för någon annan klad (data visas inte).
Koalescenta artträdsanalyser av SNP:er. De två SNP-baserade artträdskörningarna i SNAPP konvergerade, och resultatdata kombinerades, och producerade ett slutgiltigt set av 15 639 träd. De koalescenta analyserna av RAD-SNP-datasetet producerade artträd vilka till övervägande del passar ihop med topologin av *BEAST-artträdet av Sangerdatan (se Fig. 2b). Samma större klader är återfunna med högt stöd, ehuru inom kladen B3 är El Hierro identifierat som basalt men den följande ordningen av divergens är olöst, och divergensen inom klad B4 är olöst. Artspecifika grenlängder är korta, med anledning av borttagandet av de flesta autapomorfier.
Diskussion
Oberoende av metod och dataset visar vår data att de afrokanariska blåmesarna är monofyletiska och representerar fyra större klader. Det afrokanariska komplexet har ett kontinentalt ursprung, och Kanarieöarna har koloniserats tre gånger (Fig. 2). Populationerna på La Palma och i Libyen representerar kvarlevor av en nordafrikansk släktpopulation, antingen med delad gemensam förfader separerade från de andra afrokanariska ättlingarna (Fig. 2) eller särskilda i hastig sekvens från den afrikanska stamfadern (Fig. 4 och S5, Stödinformation).
Det har förekommit mycket debatt om praxisen av sammanlänkning i supermatriser kontra koalescensbaserade artträdsmetoder, vilka integrerar artträden över markörträd (se t.ex. Gatesy & Springer 2013; McVay & Carstens 2013). Här använde vi båda tillvägagångssätten, såväl som koalescensbaserad artträdsintegration utan inferens från markörträd (Bryant et al2012). Erhållna resultat med de olika typerna av dataset (dvs. färre långa Sangersekvenser kontra många korta RAD-sekvenser, och SNP:er härledda därom) producerade konsekventa resultat med endast små skillnader (huvudsakligen rörande det exakta förhållandet mellan populationer från La Palma och Libyen; Fig. 2 och 3).
Användningen av RAD-sekvensering för interspecifika fylogenomikstudier är fortfarande sällsynt, och typiskt nog har antingen fylogenetiska analyser av sekvenssupermatriser (Eaton & Ree 2013; Wagner et al. 2013; Cruaud et al2014), SNP-supermatriser (Jones et al. 2013) eller koalescensarts-SNP-träd (Gohli et al. 2015) genomförts, men utan tvärmetodologisk utvärdering. Cruaud et al. (2014) undersökte även skillnaderna och likheterna i jämförelse med traditionell Sangersekvensering av skalbaggs-dna. Nyligen genomförda simuleringar av RAD-sekvensdata visar, att om man sammanlänkar markörer och tillåter en substantiell proportion av saknad data – och därmed ökar antalet inkluderade loci – resulterar det i mer precisa träd (Huang & Knowles 2014). Vi märker emellertid liten skillnad mellan dataset med respektive utan tolerans för saknad data (Fig. 4 och S5, Stödinformation). För förekommande skillnader avviker supermatrisen RELAXED, vilken inkluderar saknad data (Fig. 4), mer från de koalescenta artträden (Fig. 2) än supermatrisen STRICT, vilken inte inkluderar saknad data.
Bland de afrokanariska blåmesarna finns, i enlighet med Dietzen et al. (2008), Illera et al. (2011) och Päckert et al. (2013), en klad med starkt och enhetligt stöd innefattande de nordvästafrikanska populationerna och alla Kanarieöarna utom La Palma (Fig. 2 och 4). Detta delas vidare in i en ytlig klad innefattande östra Kanarieöarna och Nordvästafrika (klad B4), och en klad omfattande centrala Kanarieöarna (klad B3). Den korta grenlängden som erhållits inom klad B4 påvisar att det finns en liten differentiering bland dessa populationer, och att ömsesidig monofyli faktiskt inte förekommer mellan de olika populationerna i ett mitokondriellt markörträd (Fig. S1, Stödinformation). Detta överensstämmer med den ringa fjäderdräktsdifferentieringen mellan populationerna på Lanzarote och Furteventura (ssp. degener) jämfört med nordvästafrikanska populationer (ssp. ultramarinus) (Martin 1991); strukturella skillnader med kortare vingar och större näbbar och ben förekommer emellertid (Grant 1979). Klad B3 visar markerad differentiering, med långa grenar, basal och otvetydig position av El Hierro-populationen (ssp. ombriosus), vilket även kännetecknas av dess grönaktiga snarare än gråaktiga rygg (Martin 1991), följt av sekventiella delningar (Fig. 2 och 4). De flesta analyser stöder delningssekvensen El Hierro–Gran Canaria–La Gomera/Teneriffa (Fig. 2 och 4), men vissa konflikter visade sig i sammanlänkningsanalysen av supermatrisen RELAXED, vilken inkluderar saknad data (Fig. 4). Denna sekvens är i enlighet med gällande taxonomi med ssp. hedwigae på Gran Canaria, och ssp. teneriffaepå Teneriffa och La Gomera (Dietzen et al. 2008; Gill & Donsker 2014).
Basal för huvudkladen av de kontinentalafrikanska och insulära populationerna var populationerna från La Palma (ssp. palmensis), som kännetecknas av vitaktig snarare än gulaktig mage (Martin 1991), och smalare näbb än andra populationer på Kanarieöarna (Grant 1979), och Libyen (ssp. cyrenaicae). Dessa placerades antingen som systertaxa (koalescensbaserade artträd; Fig. 2), som upprättat av Päckert et al. (2013), eller självständig avgrening i hastig sekvens från gemensam afrokanarisk blåmesstamfader (sammanlänkningsbaserade fylogenetiska analyser; Fig. 4 och S5, Stödinformation). Gohli et al. (2015) rekommenderade att populationen på la Palma skulle tilldelas artstatus, baserat på differentiering i spermielängd (Gohli et al. 2015), i kombination med tidigare etablerad långtidsisolering (Päckert et al. 2013), och morfologisk (Grant 1979; Martin 1991) och vokal (Schottler 1993, 1995) differentiering från andra öpopulationer. Medan detta kan rättfärdigas, avstår vi från att föreslå taxonomisk revision av enstaka populationer, och rekommenderar omfattande analyser av samtliga populationer med hjälp av Bayesianska artavgränsningsmetoder (Jones et al. 2014; Yang & Rannala 2014).
Kolonisation av Kanarieöarna
Generellt betraktas tättingar på Kanarieöarna som bofasta, det vill säga det finns inga observerade fall av spridning bland öarna (Martín & Lorenzo 2011), och genetisk data stöder detta påstående (Illera et al. 2012). I själva verket finns det, för fågelarter som förekommer på flertalet öar, åtskilliga exempel som representerar mångfaldigt självständiga kolonisationstillfällen från det kontinentala fastlandet, till exempel Erithacus rubecula(Dietzen et al. 2003), Regulusspp. (Päckert et al. 2006), Lanius meridionalis (Padilla et al. 2015) och Fringillaspp. (Marshall & Baker 1999).
Baserat på morfologi föreslog Grant (1979) en enda kolonisation från fastlandet, följt av ett scenario med västlig stegvis kolonisation. Kvist et al. (2005) och Dietzen et al. (2008) föreslog en initial kolonisation av Teneriffa, från vilken andra öar koloniserades (utom La Palma, som Diezen et al. (2008) föreslog koloniserades separat från Europa). Illera et al. (2001) och Päckert et al. (2013) erkände La Palma som ett unikt kolonisationstillfälle, men drog slutsatsen att alla andra öpopulationer härstammar från ett enda kolonisationstillfälle till de centrala öarna, följt av en östlig stegvis kolonisation.
Våra trädtopologier motbevisar ovanstående modeller, och föreslår, i överensstämmelse med Gohli et al. (2015), inte mindre än tre kolonisationstillfällen. Det första representerar populationen på La Palma, vilken – precis som den libyska populationen – avknoppades från en ursprunglig afrokanarisk blåmes. Utbredningen av skogsvegetation har varierat avsevärt med de nordafrikanska monsuncyklerna över de senaste 8 miljoner år (Larrasoana et al. 2003, 2013). I cykler om 50–400 tusenårsintervall, utvecklades “gröna Saharaperioder” typiskt över 2–3 tusen år (Ky), varade 4–8 Ky och försvann över 2–3 Ky, under vilka vegetationen hastigt fragmenterades (Larrasoana et al. 2013, 2013). Sådana förändringar ger skäl till att anta att afrikanska blåmesar periodvis har varit brett utspridda i Nordafrika, men dragit sig tillbaka under torrperioder parallellt med försvinnande skogstäcke. Efter ett sådant tillbakadragande förblev den libyska populationen isolerad som en kvarleva. Klimatdriven skogsfragmentering följt av isolering har även föreslagits som en pådrivare för diversifiering bland afrikanska skogsrödhakar (inom Muscicapidae) under sen Pliocene (Voelker et al. 2010).
Det äldsta kolonisationstillfället ägde rum på La Palma, med en beräknad ålder på 1,7–2,0 miljoner år (Guillou et al. 2001; Carracedo & Day 2022), vilket skulle kunna innebära en övre gräns för divergenstillfället (jfr Datering med kalibrering C, i vilken La Palma-populationen inskränktes till en maximumålder på 3,5 miljoner år, i enlighet med Päckert et al. (2013); Tabell 1). Den genetiska differentieringen mellan de afrokanariska populationerna är omfångsrik, och datering, som inte införlivar öåldrar, beräknar nodåldrar avsevärt högre än denna gräns (3,7–4,3 miljoner år för relaxerad kalibrering A och 4,3–5,4 miljoner år för strikt kalibrering B; Tabell 1 och S5, Stödinformation; cf. Päckert et al. (2013)). Detta skulle kunna föreslå en mycket accelererad molekylär hastighet. Alternativt nådde det första kolonisationstillfället inledningsvis (en) äldre ö(ar) än La Palma (till exempel är La Gomera och Teneriffa mer än 10 miljoner år gamla). Efter dess uppkomst koloniserades sedan La Palma från en av dessa äldre öar, och populationen stannade kvar där, medan populationer på andra öar utplånades eller blev utkonkurrerade av en andra kolonisation från fastlandet. Ytterligare ett alternativ skulle kunna vara att differentieringen av La Palma-ättlingarna ägde rum på fastlandet före uppkomsten av La Palma. Därefter skulle La Palma ha koloniserats, medan källpopulationen skulle ha utrotats.
Vår data infererar att en andra kolonisation (eller tredje; se diskussionen nedan om kladåldrar) inträffade från det afrikanska fastlandet till centrala Kanarieöarna. Populationen på El Hierro utgörs av den basala singelgrenen, men vi menar att El Hierro förmodligen inte är målet för den första kolonisationen och en källa för efterföljande östlig stegvis kolonisation av de återstående centrala öarna. Vi baserar detta påstående på tre omständigheter: (i) Öns ålder beräknas vara endast 1,2 miljoner år (Carracedo & Day 2002), att jämföras med en beräknad basal nodålder (B3) på 1,5–2,2 miljoner år för relaxerad kalibrering A och 1,8–2,7 miljoner år för strikt kalibrering B (Tabell 1 och S5, Stödinformation). (ii) Populationen på El Hierro uppvisar mycket låg gällande genetisk diversitet, vilket är fyr- till sexfaldigt lägre än den för populationerna på de andra öarna (Tabell S5, Stödinformation; se även Illera et al. 2011). (iii) Den låga genetiska diversiteten verkar inte härröra från någon nyligen förekommen flaskhals, då den demografiska rekonstruktionen för El Hierro visar en lång tids låg populationsstorlek, vilken har ökat något på senare tid (Fig. 3c och S3c, Stödinformation).
En enkel bana för stegvis kolonisation från El Hierro är således inte rimlig. Vi kan endast spekulera i den exakta banan, men våra resultat torde förorda ett scenario där en inledande kolonisation av en av de större öarna, till exempel Gran Canaria, följdes av självständiga kolonisationer därifrån till El Hierro (möjligtvis av en annan ö, från vilken populationen senare utrotades eller utkonkurrerades) och Teneriffa, och från Teneriffa vidare till La Gomera. Alternativt skulle Teneriffa, i analogi med delar av förslagen av Kvist et al. (2005), kunna ha varit den första ön att bli koloniserad, från vilken samtliga andra centrala öar koloniserades: först El Hierro (möjligtvis via t.ex. La Gomera, från vilken populationen senare utrotades eller utkonkurrerades), sedan Gran Canaria och till sist La Gomera (en (åter)kolonisation som skulle ersätta tidigare restpopulationer). Sådana scenarier är, i strid med vanliga intuitiva tolkningar, fullständigt kompatibla med artträdstopologin (Fig. 2), och vi skulle vilja hävda att de representerar återhållsamma tolkningar baserade på trädtopologi, genetisk diversitet (Tabell S3, S7 och S8, Stödinformation), demografisk historia (Fig. 3) och geologisk historia av öarna.
Den tredje kolonisationen (eller den andra; se diskussionen nedan om kladåldrar) nådde de östra öarna Fuerteventura och Lanzarote. Det finns en märkbar skillnad mellan artträdsmetoder, i vilka klad B4 är mycket ytlig (103 Ky, 95 % HPD 66–149 Ky; Fig. 2), och RAD-baserade sammanlänkningsanalyser, i vilka kladerna är daterade till 1,3–2,9 miljoner år (Tabell 1 och S5, Stödinformation). Upptäckten av flaskhalsar i de östra kanarieöpopulationerna, kompatibla med typiska effekter av få grundarindivider, beräknas också vara mycket nyligen inträffade (27,5 Kya; Fig. 3a), mer i överensstämmelse med artträdsanalyserna. Således kommer koalescensanalyser, baserade på Sangerdata och applicerade med en strikt molekylär klocka, fram till en senare klad B4, medan kalibreringspunktsbaserade fylogenetiska analyser av sammanlänkad RAD-sekvenseringsdata, med en relaxerad molekylär klocka, producerar en avsevärt högre beräknad ålder. Det finns indikationer på att skillnaden mellan koalescensartträdsmetoder och sammanlänkningsanalyser kan återskapas: att döma från relativa nodåldrar, kom även Gohli et al. (2015) fram till en senare klad B4 baserat på koalescensartträdsanalyser av SNP:er, medan fylogenetisk datering av sammanlänkad sekvensdata hos Päckert et al. (2013) genererade nodåldrar av c. 1,4–2,7 miljoner år (beroende på huruvida en Medelhavsöpopulation inkluderades). I själva verket beräknas klad B4 vara marginellt äldre än klad B3 (Tabell 1 och S5, Stödinformation), när El Hierros och La Palmas vulkaniska åldrar inte appliceras som restriktioner.
Det verkar som om anledningen till den drastiskt lägre RAD-sekvenseringsframgången med proverna från Lanzarote och Teneriffa (Tabell S4, Stödinformation) kan vara generellt lägre DNA-kvalitet. Den jämförbart mycket stora genetiska variationen inom populationerna (individerna) från Lanzarote och Teneriffa (Fig. S5, Stödinformation) reflekterar inte den mycket låga genetiska diversiteten i de populationerna (Tabell S3, S7 och S8, Stödinformation); likaledes avviker separationen av Lanzaroteprovet från återstående klad B4 och Teneriffaprovet från klad B3 (Fig. S5 och S6, Stödinformation), från de populationsbaserade resultaten (Fig. 2a). Detta antyder att den lägre DNA-kvaliteten manifesteras i sekvenseringsresultaten som falsk genetisk diversitet. Trots att vi är osäkra på av vilken mekanism sekvenseringsfel kan ha introducerats, betraktar vi dateringsresultaten där de proverna är uteslutna som mer tillförlitliga.
Generellt introducerar kalibrering utan bra fossilinformation stor osäkerhet, och variationen i våra dateringsresultat är i paritet med nyligen daterade fylogenier (Päckert et al. 2013). Det är värt att poängtera att 95 % högsta efterföljande densitetsintervall är stora och överlappar mellan olika inställningar för dateringsanalyserna (Tabell S5, Stödinformation). Den bästa tillämpningen av transformering av geologiska händelser till tillförlitliga kalibreringspunkter kan också diskuteras. I fallet med salthaltskrisen i sena miocen (SKM), använde Päckert et al. (2013) tiden för uttorkningen av Medelhavet som ett övre gräns för divergensen mellan europeiska och nordafrikanska svartmesättlingar (men utan någon lägre gräns). Medan vi håller med om att det inte är sannolikt att det kan ha funnits begränsat genflöde mellan ättlingarna även efter att Medelhavet fyllts på, skulle det huvudsakliga delningstillfället ha inträffat samtidigt med SKM. Vi inkluderade därför dateringsanalyser i vilka strikta ingångsvärden, med täckning för endast uttorkningsperioden, tillämpades för SKM (kalibrering B, och även D och E, vilka är liknande; Tabell 1 och S5, Stödinformation).
Vilka är käll- och målpopulationerna?
Öpopulationer är ofta betraktade som underordnade dem på fastlandet och implicit antagna för att vara resultatet av kolonisation från fastlandskällor. Fastän detta förmodligen är sant i de flesta fall, har detta synsätt utmanats på senare tid, och det finns nu exempel där öpopulationer kan ha utgjort källa för omvänd fastlandskolonisation (Filardi & Moyle 2005; Bellemain et al. 2008; Jønsson et al. 2011). Även om de är sällsynta eller åtminstone inte vanligt förekommandeär dylika återkolonisationer, från ö till fastland, viktiga att dokumentera då de skulle stödja öars potentiella roll som källa för fastlandssystem som motsats till den traditionella synen på öar som enkelriktade evolutionsspår (kolonisationsmål). Återkolonisationsdemografi bildar underlag för både grundarexpansions- (founder-flush) och den peripatriska artbildningsmodellen artbildningsmodellen (Mayr 1963; Powell 1978; Slatkin 1996), modeller som har kritiserats som högst osannolika (Charlesworth & Smith 1982; Barton & Charlesworth 1984; Charlesworth 1997).
I den afrokanariska blåmesens fall antyder resultaten i Illera et al. (2011) och Päckert et al. (2013) en sådan återkolonisation från de små östra öpopulationerna till de stora fastlandspopulationerna i Marocko och Algeriet, men detta ges inte stöd av våra data. I stället fann vi (i) låga nivåer av genetisk variation i öpopulationerna, i kontrast till hög fastställd variation på Afrikas fastland (Tabell S3, S7 och S8, Stödinformation); detta observeras även i ett senare mikrosatellitdataset (Hansson et al. 2014). Illera et al. (2011) rapporterar relativt högre genetisk diversiteti Kanarieöpopulationerna än i Nordafrika, men detta verkar ha en stark mitokondriell grund, då kärngenen visar det motsatta mönstret. Vidare (ii) sätter våra trädbaserade analyser fastlandspopulationerna basalt i förhållande till populationerna på de östra Kanarieöarna (Fig. 2, 4 och S5, Stödinformation), i en systerklad till den som omfattar de centrala Kanarieöarna. Detta är i enlighet med Gohli et al. (2015), men skiljer sig från positionen för populationerna i Nordafrika och på de östra öarna, som av Päckert et al.(2013) rapporterades som inneslutna i samma klad som de centrala öarnas populationer. Slutligen (iii) visar våra demografiska rekonstruktioner tydliga förtecken på mycket färska starka flaskhalsar för populationerna på Fuerteventura och Lanzarote (Fig. 3b), som är ett tecken kompatibelt med nyligen inträffat grundande av dessa populationer, medan inga dylika mönster kan urskiljas för fastlandspopulationerna (Fig. 3a). Således ger våra data inte stöd åt att de för närvarande relativt stora fastlandspopulationerna har utvidgats hastigt efter att ha uppstått från de östra Kanarieöarna.
Slutsats
Den afrokanariska blåmesens evolutionshistoria har varit fortsatt omdebatterad trots intensiva försök att lösa den (Salzburger et al. 2002; Kvist et al. 2005; Dietzen et al. 2008; Illera et al. 2011; Päckert et al. 2013; Hansson et al. 2014; Gohli et al. 2015). Enkellocusanalyser och sammanlänkningsanalyser av flera locus, baserade på endast ett fåtal gener, med en övervikt mot mitokondriella markörer, har producerat oförenliga resultat. Dessutom begränsades en nyligen gjord SNP-baserad studie (Gohli et al. 2015) av avvikande provtagning av en nyckelpopulation, beskrivna förhållanden bland grupper såväl som populationsdemografi med låg effektiv provstorlek, och avsaknad av nodalt stöd för de djupare noderna, vilket resulterar i osäkra evolutionstolkningar. Här förenade vi omfångsrik traditionell sekvensering och parallellsekvensering och påvisade att Kanarieöarna har blivit koloniserat från det afrikanska fastlandet vid tre separata tillfällen, efter vilka substantiell differentiering har förekommit. De två perifera populationerna på La Palma respektive i Libyen, utgör kvarlevor av en population som samtliga afrokanariska taxa härstammar från. Vidare bestrider vår studie starkt återkolonisation från Kanarieöarna till det afrikanska fastlandet och ansluter sig i stället till den traditionella synen på öar som mestadels kolonisationsmål. Baserat på trädtopologin, återvunnen av Illera et al. (2011) och Päckert et al. (2013), kan deras tolkning verifieras, att Nordvästafrika återkoloniserades från Kanarieöarna. Vi hävdar emellertid att deras analyser var bristfälliga eftersom de inkluderade mycket få kärnloci, och eftersom ofullständig härkomstssortering kan ha försvagat verkliga mönster. För att lösa sådan komplex evolutionshistoria hävdar vi att stor genomisk täckning såväl som tät och jämförbar taxonprovtagning är nödvändig och att koalescensbaserade metoder för artträdsinferens, vilket är mest passande för populationsprover av flera individer, rutinmässigt skulle undersökas. Därför borde parallellsekvensering med reducerad representation i det förestående spela en viktig roll i fylogeografi.
Tack
Vi tackar Jens Hering och Martin Päckert, Senckenberg naturhistoriska samling, för tillhandahållande av prover från C. t. cyrenaicaefrån Libyen ochPeripausspp.; Andrey Gavrilov, Chokpak fågelstation, och Mats Waern för tillhandahållande av C. cyanus-prover från Kazakstan; José Luis Copete och Ferran López för tillhandahållande av prover av P. a. atlas från Marocko; CHAGRA ringmärkningsgrupp för tillhandahållande av C. teneriffae-prover från Ceuta; Barboutis Christos för tillhandahållande av C. caeruleus-prover från Kreta; Matt Wood och Edward Grey-institutet vid Oxford för tillhandahållande av prover från C. c. obscurus; Olof Hellgren och Kvismare fågelstation för tillhandahållande av prover från Parus major och Pœcile palustris. Martí Franch tillhandahöll illustrationerna på Cyanistes. Den regionala regeringen på Kanarieöarna, Andalusien och Madrid i Spanien och regeringen i Marocko gav tillstånd att utföra fältarbete i dessa länder. Vi vill tacka två anonyma textgranskare för värdefulla kommentarer på manuskriptet. M.S. önskar tacka the forskningsskolan Geneco för träning och stöd. Denna studie genomfördes med stöd från Svenska vetenskapsrådet (621-2007-5381 och 621-2009-4945), Stiftelsen Oscar och Lili Lamms Minne, Crafoordska stiftelsen, EU-FP7 (projekt Fågelgenomik) (samtligt till B.H.) och Kungliga Fysiografiska Sällskapet (till M.S.).
Referenser
Baird NA, Etter PD, Atwood TS, et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3, e3376.
Balke M, Ribera I, Hendrich L et al. (2009) New Guinea highland origin of a widespread arthropod supertramp. Proceedings of the Royal Society B-Biological Sciences, 276, 2359–2367.
Barton NH, Charlesworth B (1984) Genetic revolutions, founder effects, and speciation. Annual Review of Ecology and Systematics, 15, 133–164.
Bellemain E, Ricklefs RE (2008) Are islands the end of the colonization road? Trends in Ecology & Evolution, 23, 461–468.
Bellemain E, Bermingham E, Ricklefs RE (2008) The dynamic evolutionary history of the bananaquit (Coereba flaveola) in the Caribbean revealed by a multigene analysis. BMC Evolutionary Biology, 8, 240.
BirdLife International, NatureServe (2014) Bird Species Distribution Maps of the World. BirdLife International, Cambridge.
Bouckaert R, Heled J, Kuhnert D et al. (2014) BEAST 2: a software platform for bayesian evolutionary analysis. Plos Computational Biology, 10, e1003537.
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Molecular Biology and Evolution, 29, 1917–1932.
Carracedo JC, Day S (2002) Canary Islands. Terra Publishing, Hertfordshire, London.
Carrascal LM, Moreno E, Valido A (1994) Morphological evolution and changes in foraging behavior of island and mainland populations of Blue Tit (Parus-Caeruleus) – a test of convergence and ecomorphological hypotheses. Evolutionary Ecology, 8, 25–35.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) STACKS: an analysis tool set for population genomics. Molecular Ecology, 22, 3124–3140.
Charlesworth B (1997) Is founder-flush speciation defensible? American Naturalist, 149, 600–603.
Charlesworth B, Smith DB (1982) A computer-model of speciation by founder effects. Genetical Research, 39, 227–236.
Corl A, Ellegren H (2013) Sampling strategies for species trees: the effects on phylogenetic inference of the number of genes, number of individuals, and whether loci are mitochondrial, sex-linked, or autosomal. Molecular Phylogenetics and Evolution, 67, 358–366.
Cramp S, Perrins C (1993) Handbook of the Birds of Europe, the Middle East, and North Africa: The Birds of the Western Palearctic. Oxford University Press, Oxford.
Cruaud A, Gautier M, Galan M et al. (2014) Empirical assessment of RAD sequencing for interspecific phylogeny. Molecular Biology and Evolution, 31, 1272–1274.
Danecek P, Auton A, Abecasis G et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158.
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9, 772.
Davey JW, Hohenlohe PA, Etter PD, et al. (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 12, 499–510.
Dietzen C, Witt H-H, Wink M (2003) The phylogeographic differentiation of the European robin Erithacus rubecula on the Canary Islands revealed by mitochondrial DNA sequence data and morphometrics: evidence for a new robin taxon on Gran Canaria? Avian Science, 3, 115–131.
Dietzen C, Garcia-del-Rey E, Castro GD, Wink M (2008) Phylogeography of the blue tit (Parus teneriffae-group) on the Canary Islands based on mitochondrial DNA sequence data and morphometrics. Journal of Ornithology, 149, 1–12.
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. Plos Biology, 4, 699–710.
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973.
Eaton DAR, Ree RH (2013) Inferring phylogeny and introgression using RADseq Data: an example from flowering plants (Pedicularis: Orobanchaceae). Systematic Biology, 62, 689–706.
Ekblom R, Galindo J (2011) Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity (Edinb), 107, 1–15.
Ellegren H (2007) Molecular evolutionary genomics of birds. Cytogenetic and Genome Research, 117, 120–130.
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA (2011) SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods in Molecular Biology, 772, 157–178.
Fern_andez-Palacios JM, de Nascimento L, Otto R et al. (2011) A reconstruction of Palaeo-Macaronesia, with particular reference to the long-term biogeography of the Atlantic island laurel forests. Journal of Biogeography, 38, 226–246.
Filardi CE, Moyle RG (2005) Single origin of a pan-Pacific bird group and upstream colonization of Australasia. Nature, 438, 216–219.
Gatesy J, Springer MS (2013) Concatenation versus coalescence versus “concatalescence”. Proceedings of the National Academy of Sciences, USA, 110, E1179.
Gill FB, Donsker D (2014) IOC World Bird List (v 4.3). Available from: http://www.worldbirdnames.org/.
Gillespie RG, Croom HB, Palumbi SR (1994) Multiple origins of a spider radiation in Hawaii. Proceedings of the National Academy of Sciences, USA, 91, 2290–2294.
Gladenkov AY, Oleinik AE, Marincovich L, Barinov KB (2002) A refined age for the earliest opening of Bering Strait. Palaeogeography Palaeoclimatology Palaeoecology, 183, 321–328.
Gohli J, Leder EH, Garcia-del-Rey E et al. (2015) The evolutionary history of Afrocanarian blue tits inferred from genomewide SNPs. Molecular Ecology, 24, 180–191.
Gosler A, Clement P (2007) Family Paridae (tits and chickadees). In: Handbook of the Birds of the World: Vol. 12: Picathartes to Tits and Chickadees (eds del Hoyo J, Elliott A, Christie DA), pp. 662–750. Lynx Edicions, Barcelona.
Grant PR (1979) Ecological and morphological variation of Canary Island blue tits, Parus-Caeruleus (Aves, Paridae). Biological Journal of the Linnean Society, 11, 103–129.
Grant PR, Grant BR (2011) How and Why Species Multiply The Radiation of Darwin’s Finches. Princeton University Press, Princeton, New Jersey.
Guillou H, Carracedo JC, Duncan RA (2001) K-Ar, Ar-40-Ar-39 ages and magnetostratigraphy of Brunhes and Matuyama lava sequences from La Palma Island. Journal of Volcanology and Geothermal Research, 106, 175–194.
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704.
Hansson B, Ljungqvist M, Illera JC, Kvist L (2014) Pronounced fixation, strong population differentiation and complex population history in the Canary Islands blue tit subspecies complex. PLoS ONE, 9, e90186.
Heled J, Drummond AJ (2008) Bayesian inference of population size history from multiple loci. BMC Evolutionary Biology, 8, 289.
Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27, 570–580.
Huang H, Knowles LL (2014) Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Systematic Biology. doi: 10.1093/sysbio/syu046.
Illera JC, Koivula K, Broggi J et al. (2011) A multi-gene approach reveals a complex evolutionary history in the Cyanistes species group. Molecular Ecology, 20, 4123–4139.
Illera JC, Rando JC, Richardson DS, Emerson BC (2012) Age, origins and extinctions of the avifauna of Macaronesia: a synthesis of phylogenetic and fossil information. Quaternary Science Reviews, 50, 14–22.
Johansson US, Ekman J, Bowie RC et al. (2013) A complete multilocus species phylogeny of the tits and chickadees (Aves: Paridae). Molecular Phylogenetics and Evolution, 69, 852–860.
Jones JC, Fan SH, Franchini P, Schartl M, Meyer A (2013) The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome-wide approach using restriction site-associated DNA sequencing. Molecular Ecology, 22, 2986–3001.
Jones G, Aydin Z, Oxelman B (2014) DISSECT: an assignmentfree Bayesian discovery method for species delimitation under the multispecies coalescent. Bioinformatics. doi: 10.1093/bioinformatics/btu770.
Jønsson KA, Bowie RCK, Moyle RG et al. (2010) Historical biogeography of an Indo-Pacific passerine bird family (Pachycephalidae): different colonization patterns in the Indonesian and Melanesian archipelagos. Journal of Biogeography, 37, 245–257.
Jønsson KA, Fabre PH, Ricklefs RE, Fjeldsa J (2011) Major global radiation of corvoid birds originated in the proto-Papuan archipelago. Proceedings of the National Academy of Sciences, USA, 108, 2328–2333.
Juan C, Emerson BC, Oromi P, Hewitt GM (2000) Colonization and diversification: towards a phylogeographic synthesis for the Canary Islands. Trends in Ecology & Evolution, 15, 104–109.
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780.
Krijgsman W, Hilgen FJ, Raffi I, Sierro FJ, Wilson DS (1999) Chronology, causes and progression of the Messinian salinity crisis. Nature, 400, 652–655.
Kvist L, Broggi J, Illera JC, Koivula K (2005) Colonisation and diversification of the blue tits (Parus caeruleus teneriffae-group) in the Canary Islands. Molecular Phylogenetics and Evolution, 34, 501–511.
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nature Methods, 9, 357–359. Larrasoana JC, Roberts AP, Rohling EJ, Winklhofer M,
Wehausen R (2003) Three million years of monsoon variability over the northern Sahara. Climate Dynamics, 21, 689–698.
Larrasoana JC, Roberts AP, Rohling EJ (2013) Dynamics of green Sahara periods and their role in hominin evolution. PLoS ONE, 8, e76514.
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452.
Losos JB (2011) Lizards in an Evolutionary Tree: Ecology and Adaptive Radiation of Anoles. University of California Press, Oakland, California.
Marincovich L, Gladenkov AY (1999) Evidence for an early opening of the Bering Strait. Nature, 397, 149–151.
Marshall HD, Baker AJ (1999) Colonization history of Atlantic island common chaffinches (Fringilla coelebs) revealed by mitochondrial DNA. Molecular Phylogenetics and Evolution, 11, 201–212.
Martin JL (1991) Patterns and significance of geographical variation in the Blue Tit (Parus caeruleus). Auk, 108, 820–832.
Mart_ın A, Lorenzo JA (2001) Aves del archipi_elago canario. Francisco Lemus Editor, San Cristobal De La Laguna.
Mayr E (1963) Animal Species and Evolution. Harvard University Press, Cambridge, Massachusetts.
McVay JD, Carstens BC (2013) Phylogenetic model choice: justifying a species tree or concatenation analysis. Journal ofPhylogenetics and Evolutionary Biology, 1, 114.
Medail F, Quezel P (1999) Biodiversity hotspots in the Mediterranean basin: setting global conservation priorities. Conservation Biology, 13, 1510–1513.
Melo M, Warren BH, Jones PJ (2011) Rapid parallel evolution of aberrant traits in the diversification of the Gulf of Guinea whiteeyes (Aves, Zosteropidae). Molecular Ecology, 20, 4953–4967.
Päckert M, Dietzen C, Martens J, Wink M, Kvist L (2006) Radiation of Atlantic goldcrests Regulus regulus spp.: evidence of a new taxon from the Canary Islands. Journal of Avian Biology, 37, 364–380.
Päckert M, Martens J, Tietze DT et al. (2007) Calibration of a molecular clock in tits (Paridae)–do nucleotide substitution rates of mitochondrial genes deviate from the 2% rule? Molecular Phylogenetics and Evolution, 44, 1–14.
Päckert M, Martens J, Sun YH et al. (2011) Horizontal and elevational phylogeographic patterns of Himalayan and South-east Asian forest passerines (Aves: Passeriformes). Journal of Biogeography, 39, 556–573.
Päckert M, Martens J, Hering J, Kvist L, Illera JC (2013) Return flight to the Canary Islands–the key role of peripheral populations of Afrocanarian blue tits (Aves: Cyanistes teneriffae) in multi-gene reconstructions of colonization pathways. Molecular Phylogenetics and Evolution, 67, 458–467.
Padilla DP, Spurgin LG, Fairfield EA, Illera JC, Richardson DS (2015) Population history, gene flow, and bottlenecks in island populations of a secondary seed disperser, the southern grey shrike (Lanius meridionalis koenigi). Ecology and Evolution, 5, 36–45.
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics, 20, 289–290.
Partridge L, Pringmill F (1977) Canary Island Blue Tits and English coal tits – convergent evolution. Evolution, 31, 657–665.
Patel S, Kimball RT, Braun EL (2013) Error in phylogenetic estimation for bushes in the tree of life. Journal of Phylogenetics and Evolutionary Biology, 1, 110.
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A (2009) How many bootstrap replicates are necessary? Research in Computational Molecular Biology, Proceedings, 5541, 184–200.
Powell JR (1978) The founder-flush speciation theory: an experimental approach. Evolution, 32, 465–474.
Purcell S, Chang C (2014) Plink 1.9beta2c, Available from: https://www.cog-genomics.org/plink2.
R Core Team (2013) R: A Language and Environment for Statistical Computing. Available from: http://www.R-project.org
Rambaut A, Drummond AJ (2007) Tracer v1. Available from: http://tree.bio.ed.ac.uk/software/tracer/.
Salzburger W, Martens J, Sturmbauer C (2002) Paraphyly of the Blue Tit (Parus caeruleus) suggested from cytochrome b sequences. Molecular Phylogenetics and Evolution, 24, 19–25.
Sambrook J, Russel DW (2001) Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Sangster G (2006) The taxonomic status of ‘phylogroups’ in the Parus teneriffae complex (Aves): comments on the paper by Kvist et al. (2005). Molecular Phylogenetics and Evolution, 38, 288–289; author reply 290.
Savolainen V, Anstett MC, Lexer C et al. (2006) Sympatric speciation in palms on an oceanic island. Nature, 441, 210–213.
Schottler B (1993) Canary Islands Blue Tits (Parus caeruleus ssp) – differences and variation in territorial song – preliminary results. Boletim do Museu Municipal do Funchal, (Suppl 2), 273–277.
Schottler B (1995) Songs of blue tits Parus caeruleus palmensis from La Palma (Canary Islands): a test of hypotheses. Bioacoustics, 6, 135–152.
Schwarz G (1978) Estimating dimension of a model. Annals of Statistics, 6, 461–464.
Slatkin M (1996) In defense of founder-flush theories of speciation. American Naturalist, 147, 493–505.
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313.
Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. American Journal of Human Genetics, 73, 1162–1169.
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68, 978–989.
Voelker G, Outlaw RK, Bowie RCK (2010) Pliocene forest dynamics as a primary driver of African bird speciation. Global Ecology and Biogeography, 19, 111–121.
Wagner CE, Keller I, Wittwer S et al. (2013) Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Molecular Ecology, 22, 787–798.
Warren BH, Bermingham E, Bourgeois Y et al. (2012) Hybridization and Barriers to Gene Flow in an Island Bird Radiation. Evolution, 66, 1490–1505.
Warren BH, Simberloff D, Ricklefs RE et al. (2015) Islands as model systems in ecology and evolution: prospects fifty years after MacArthur-Wilson. Ecology Letters, 18, 200–217.
Weir JT, Schluter D (2008) Calibrating the avian molecular clock. Molecular Ecology, 17, 2321–2328.
Wickham H (2009) ggplot2: Elegant Graphics for Data Analysis. Springer, New York.
Yang Z, Rannala B (2014) Unguided species delimitation using DNA sequence data from multiple loci. Molecular Biology and Evolution, 31, 3125–3135.
Stödinformation
Ytterligare stödinformation återfinns på engelska i den länkade pdf-filen. Den innehåller följande material:
- Appendix S1 Metoder.
- Tabell S1 Provuppgifter och tillträdesnummer.
- Tabell S2 Markör- och prajmeruppgifter, PCR-inställningar etc.
- Tabell S3 Nukleotiddiversitetinom populationer.
- Tabell S4 Sammanfattning av resultat från restriktionspunktssassocierade (RAD) sekvenseringen.
- Tabell S5 Fullständig dateringsresultat från fem kalibreringar, två taxonset, samt fyra genomiska subset.
- Tabell S6 Substitutionsmodeller för de 20 Sangersekvenseringsmarkörerna.
- Tabell S7 Haplotypdiversitetinom populationer.
- Tabell S8 Theta inom populationer.
- Tabell S9 Sammanfattning av dateringsanalyserna, med belysning av skillnaden i beräknade nodåldrar mellan partiell och fullständig taxonset.
- Tabell S10 Sammanfattning av dateringsanalyserna, med belysning av skillnaden i beräknade nodåldrar mellan de fem olika kalibreringarna.
- Fig. S1 Fylogenetiskt träd för den mitokondriella kontrollregionen.
- Fig. S2 Koalescensbaserade artträd baserade på analyeser av populationsprover av 18 nukleära Sangersekvensmarkörer (men inte mitokondriella markörer).
- Fig. S3 Bayesianska siluettdiagramsanalyser baserade på Sangersekvensdata för flera loci, med täckning för hela tidsperioden av demografiska rekonstruktioner.
- Fig. S4 Bayesianska siluettdiagramsanalyser baserade på Sangersekvensdata med flera loci, med täckning för de senaste 500 000 åren.
- Fig. S5 Fylogenomisk maxsannolikhetsträd av supermatris STRICT.
- Fig. S6 Klusterindelning med multidimensionell skalning.
Referens till usrprungsartikeln:
Stervander M, Illera JC, Kvist L, Barbosa P, Keehnen NP, Pruisscher P, Bensch S & Hansson B. 2015. Disentangling the complex evolutionary history of the Western Palearctic blue tits (Cyanistes spp.) – phylogenomic analyses suggest radiation by multiple colonization events and subsequent isolation. Molecular Ecology 24(10): 2477–2494. DOI: 10.1111/mec.13145.